家族性良性血尿和 Alport 综合征在遗传方式、临床表现、肾脏病理、电镜表现、预后等方面存在不同 。
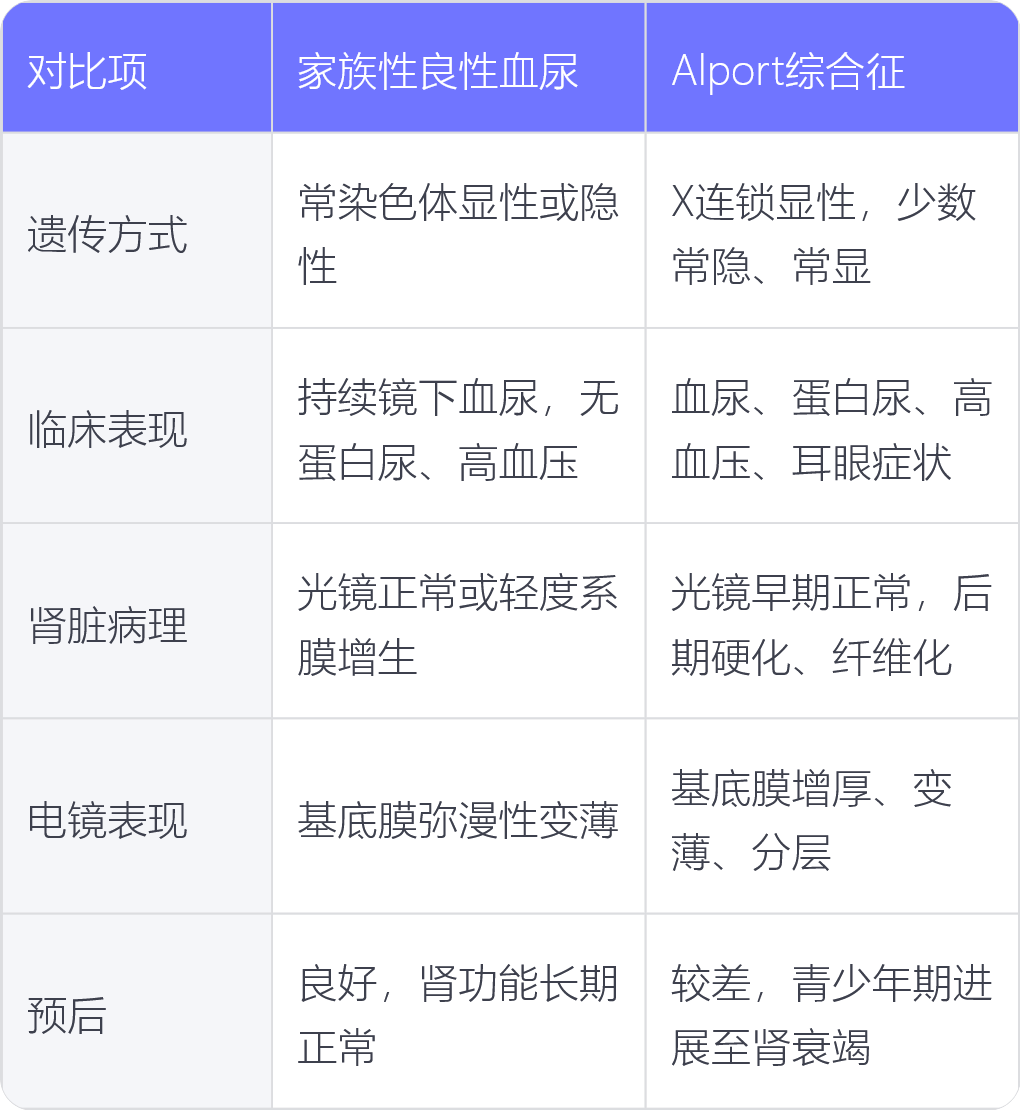
1. 遗传方式:家族性良性血尿多为常染色体显性遗传,也有常染色体隐性遗传;Alport 综合征主要为 X 连锁显性遗传,少数为常染色体隐性遗传和常染色体显性遗传。
2. 临床表现:家族性良性血尿主要表现为持续性镜下血尿,部分患者有发作性肉眼血尿,一般无蛋白尿、高血压及肾功能损害;Alport 综合征除血尿外,常伴有蛋白尿,部分患者有高血压,还可出现耳部、眼部等肾外表现,且随病情进展可出现肾功能进行性减退。
3. 肾脏病理:家族性良性血尿光镜下多正常或轻度系膜增生;Alport 综合征光镜下早期可正常,随病情进展可出现肾小球硬化、肾小管间质纤维化等。
4. 电镜表现:家族性良性血尿电镜下肾小球基底膜弥漫性变薄;Alport 综合征电镜下肾小球基底膜增厚、变薄及分层等特征性改变。
5. 预后:家族性良性血尿预后良好,多数患者肾功能可长期维持正常;Alport 综合征预后较差,多数患者在青少年或青年期进展至肾衰竭。
日常中,无论是家族性良性血尿还是 Alport 综合征患者,都应注意休息,避免劳累、感染等加重肾脏负担的因素。定期进行尿常规、肾功能等检查,以便及时发现病情变化。严格遵循医生的治疗方案,按时服药,切不可自行增减药量或停药,出现异常及时就医。